 Interview du Pr Gabriel Laurent, auteur de l’article «Mutations PRKAG2 : Haute prévalence des complications arythmiques et hémodynamiques »
Interview du Pr Gabriel Laurent, auteur de l’article «Mutations PRKAG2 : Haute prévalence des complications arythmiques et hémodynamiques »
High prevalence of arrhythmic and myocardial complications in patients with cardiac glycogenosis due to PRKAG2 mutations
Julien Thevenon, Gabriel Laurent, Flavie Ader, Pascal Laforêt, Didier Klug, Anju Duva Pentiah, Laurent Gouya, Claude Alain Maurage, Salem Kacet, Jean-Christophe Eicher, Juliette Albuisson, Michel Desnos, Eric Bieth, Denis Duboc, Laurent Martin, Patricia Réant, François Picard, Claire Bonithon-Kopp, Elodie Gautier, Christine Binquet, Christel Thauvin-Robinet, Laurence Faivre,
Europace. 2016 May 17. pii: euw067.
Les cardiomyopathies hypertrophiques (CMH) font parties d’un groupe hétérogène de maladies qui correspondent toutes à un épaississement des parois ventriculaires. En dehors de conditions anormales de la postcharge, il existe une origine génétique de ces maladies dans 40 à 60% des cas. La transmission est autosomique dominante et la plupart des gènes identifiés codent pour les protéines du sarcomère impliquées dans la performance myofibrillaire. Certaines CMH de la famille des maladies de surcharges glycogéniques et/ou de surcharge d’origine lysosomale, peuvent être associées à des préexcitation ventriculaires (PV) de type faisceau accessoires atrio-ventriculaires ou nodo-fasciculaires, mais également à des troubles de conduction atrio-ventriculaires de haut degré. On peut citer, outre la maladie de Fabry, la maladie de Pompe, la maladie de Danon, et la mutation PRKAG2. Ces anomalies de la conduction sont potentiellement dangereuses et peuvent apparaitre au fil du temps.
Le gène PRKAG2 code pour la sous unité gamma 2 de l’AMPK (5’-AMP-activated protein kinase). Les mutations de PRKAG2 sont responsables d’une accumulation du glycogène dans le tissu cardiaque, mais il est également connu que la sous unité gamma 2 de l’AMPK participe au développement embryonnaire des anneaux fibreux atrio-ventriculaires.
Depuis l’identification de cette pathologie en 2001, seulement 150 patients ont été répertoriés et c’est pourquoi nous connaissons actuellement très peu de choses sur l’évolution naturelle de cette maladie potentiellement dangereuse.
L’objectif de cette étude était de mieux définir l’histoire naturelle des CMH liées à une mutation PRKAG2, et en particulier d’identifier l’âge de survenue des complications liées aux anomalies de la conduction atrio-ventriculaire.
Patients et méthodes
Cette étude rétrospective a permis d’inclure tous les patients présentant une mutation de PRKAG2 qui ont été identifié dans le laboratoire moléculaire français des pathologies cardiaques héréditaires de l’hôpital de la Pitié-Salpêtrière entre 2001 et 2010. La séquence codante et les régions introniques flanquantes ont été séquencées par technique Sanger à partir de l’ADN génomique extrait d’un prélèvement sanguin du cas index.
Les patients ont été testé lorsqu’ils présentaient : i) une histoire familiale de mort subite cardiaque ; ii) une hypertrophie myocardique échographique (septum interventiculaire de 15 mm ou plus), iii) une CMH avec aspect de préexcitation ventriculaire (PV); iiii) un antécédent familial de PV.
Analyse statistique :
La date de survenue d’un évènement cardiovasculaire au cours du suivi a été défini pour chaque patient à partir de la date de sa naissance, il pouvait s’agir d’un trouble conductif nécessitant l’implantation d’un pacemaker, d’une mort subite cardiaque ou d’une transplantation cardiaque. Les 13 patients (4 familles) atteints de la mutation p.Arg302Gln ont été comparés aux 21 patients (5 familles) atteints d’une autre mutation.
Résultats
Une mutation de PRKAG2 a été identifiée chez 34 patients appartenant à 9 familles différentes. La rentabilité globale du test a été de 7% (0% en cas de CMH isolée et 20% en cas de PV avec ou sans CMH)
La mutation p.Arg302Gln (c.905G>A) a été identifiée dans 4/9 familles (familles 1 à 4; 13 patients). Cinq autres mutations ont été retrouvées: (p.Glu506Lys, p.Ser548Pro), dont trois nouvellement identifiées avec une haute probabilité d’être pathogène (p.Ser333Pro (c.997T>C), p.Val336Ala (c.1007T>C), et p.His530Arg (c.1589A>G).
Date de survenue d’un évènement cardiovasculaire au cours du suivi
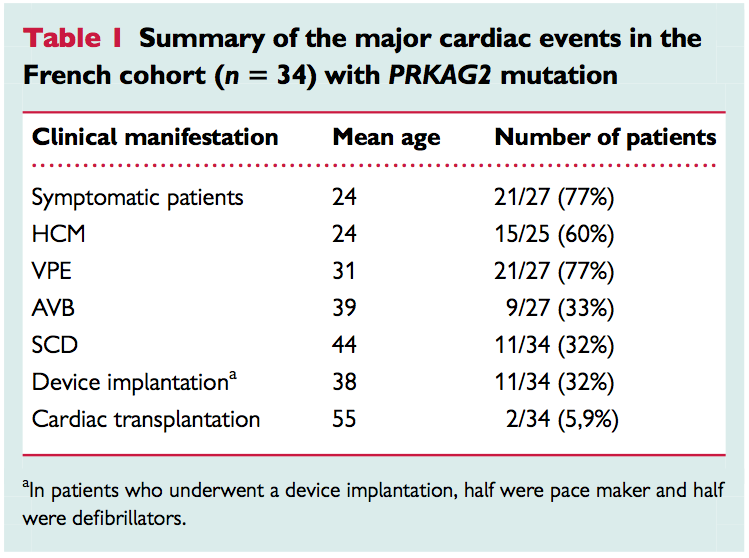
Le tableau sous-jacent résume les évènements cardiaques majeurs identifiés dans cette cohorte (HCM : Cardiomyopathie hypertrophique ; VPE : Préexcitation ventriculaire ; AVB : Bloc atrio-ventriculaire ; SCD : Décès par mort subite cardiaque ; Device implantation : pour moitié pacemaker et moitié défibrillateur implantable.
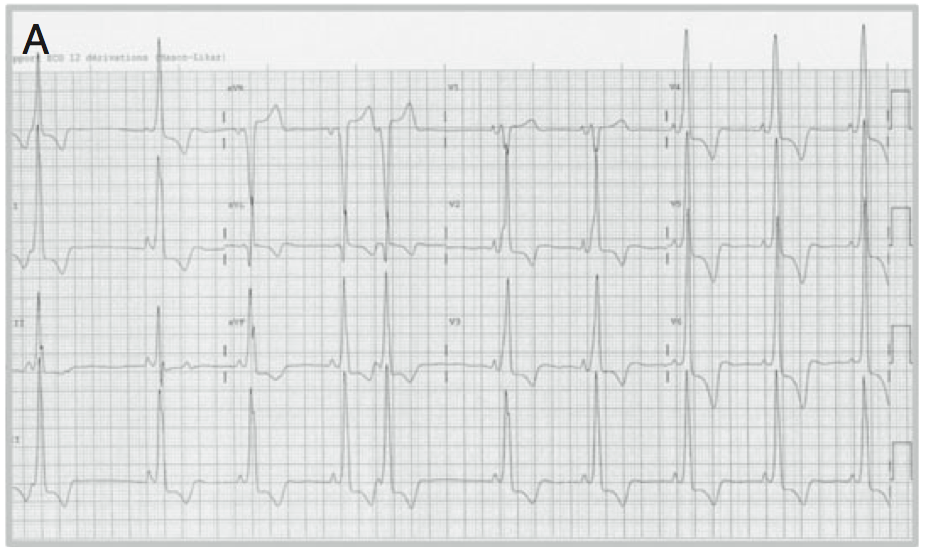
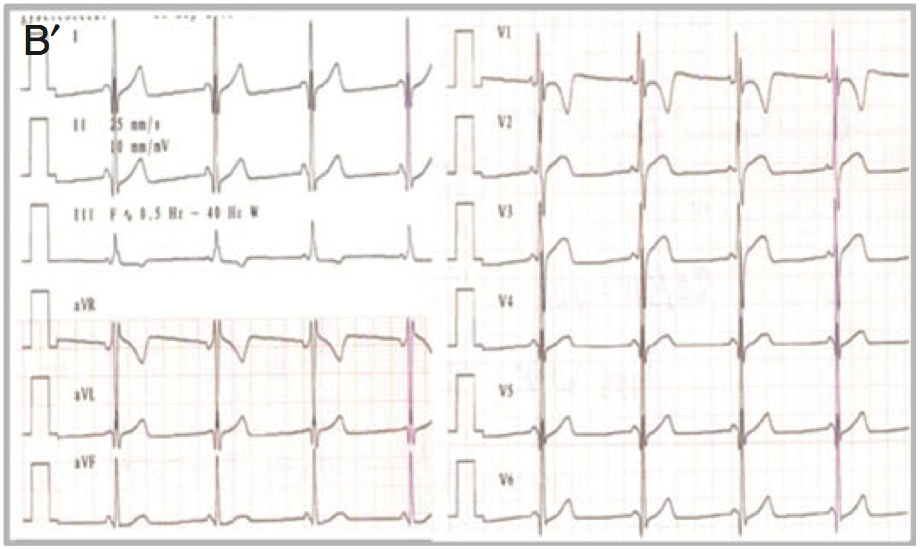
Un aspect ECG évocateur d’un syndrome de Wolf Parkinson White typique a été identifié chez 3 patients (figure A), alors que la majorité (16 patients) présentait un PR court isolé (Figure B). Comme décrits dans la littérature, des fibres de Mahaim (faisceaux nodo/fasciculo-ventriculaires) ou d’authentiques faisceaux de Kent atrio-ventriculaires ont été observés au cours d’explorations électrophysiologiques (6 patients). Aucune tachycardie jonctionnelle n’a été déclenchée mais il y a eu quelques tentatives d’ablation par radiofréquence en raison de périodes réfractaires courtes de ces voies. Tous les patients ablatés ont présentés des troubles conductifs atrio-ventriculaires dans les suites de l’ablation (sondes d’ablation en position parahissienne) nécessitant l’implantation de pacemaker.
Le risque de présenter une des expressions phénotypiques suivantes de la maladie a été évalué à l’âge de 40 ans : i) Pré excitation ventriculaire 70% (99%-CI: 50%-87%) avec un âge médian de 25 ans (IQR [20-41]); ii), CMH: 61% (99%-CI: 41%-81%) avec un âge médian de 32 ans (IQR [18-NA]) ; iii), trouble de conduction atrio-ventriculaire : 22% (99%-CI: 9%-46%) ; iiii), la survenue d’une mort subite : 20% (99%-CI: 8%-42%) ; iiiii), la probabilité d’être implanté d’un pacemaker ou d’un défibrillateur : 25% (99%-CI: 2%-48%).
Deux patients ont bénéficié d’une transplantation cardiaque à l’âge de 55 et 60 ans. L’âge médian du décès dans cette cohorte est de 60 (IQR [60-65]).
Corrélation génotype-phénotype
Aucune différence significative n’a pu être identifiée entre l’expression phénotypique, selon l’âge de survenue, des patients porteurs de la mutation p.Arg302Gln et les porteurs d’autres mutations.
Conclusions
L’association d’une préexcitation ventriculaire (aspect ECG de Wolf Parkinson et White ou PR court isolé) à une cardiomyopathie hypertrophique ; ou un contexte familial de préexcitation ventriculaire doit faire conduire à l’étude de PRKAG2.
L’expression phénotypique associée est souvent sévère tant sur le plan myocardique que rythmique avec la nécessité d’implanter un stimulateur ou un défibrillateur cardiaque dans près d’un tiers des cas de cette cohorte.
Les procédures d’ablation par radiofréquence de ces voies accessoires particulières (faisceaux para-hissiens) doivent être réalisées avec prudence compte tenu du très haut risque d’altération définitive de la conduction atrio-ventriculaire.